
Randal Tibbetts, PhD
Professor
Department of Human Oncology
I am a professor in the Department of Human Oncology (DHO) at UW–Madison, where I study genome surveillance, cell growth and neurodegeneration. Prior to joining DHO in 2010, I was an associate professor in the Department of Pharmacology at UW–Madison. My postdoctoral studies were carried out in the laboratory of Robert Abraham University (Duke University). We were among the first labs to functionally characterize members of the PI3 kinase-related kinase superfamily that includes DNA-PK, ATM and ATR—three critical regulators of the intracellular DNA damage response.
Current work in our lab is aimed at understanding how diverse growth-inhibitory signals, including DNA damage, cell-cell contact and growth factor depletion downregulate gene expression through the CREB pathway and how defects in this regulation contribute to tumorigenesis. A second related project is deciphering how alternative splicing influences DNA damage repair and tumor suppression using cellular and in vivo (mouse) models. Finally, our laboratory has developed several Drosophila melanogaster (fruit fly) models for the motor neuron disease amyotrophic lateral sclerosis. We are using these models to probe genetic mechanisms of neurodegeneration. We actively collaborate with both basic science and clinical colleagues within and outside the UW campus on each of these projects. I also teach material related to cell growth and DNA damage repair for several on-campus graduate programs, serve on both NINDS and NCI grant review panels and have served as editor and ad hoc reviewer for the Journal of Biological Chemistry, Nature, the EMBO Journal and other journals.
Education
Postdoctoral Fellow, Duke University, Cancer Biology (2000)
PhD, Northwestern University, Microbiology-Immunology (1996)
BS, University of Minnesota, Biochemistry and Chemistry (1989)
Academic Appointments
Professor, Human Oncology (2015)
Associate Professor, Human Oncology (2011)
Associate Professor, Pharmacology (2007)
Assistant Professor, Pharmacology (2001)
Selected Honors and Awards
Shaw Scientist Award, Greater Milwaukee Foundation (2003)
Robert M. and Barbara R. Bell Basic Science of Cancer Award, Duke University (2000)
Research Fellow, Leukemia Society of America (1997–2000)
National Research Service Award “Cellular and Molecular Basis of Disease” Northwestern University (1992–1994)
Stipend Recipient, Undergraduate Research Opportunities Program (UROP), University of Minnesota (1988)
American Chemical Society Outstanding Sophomore Chemistry Student (1986)
Most Outstanding Freshman Chemistry Student (1985)
Research Focus
Genome Surveillance and Cell Growth Regulation, Molecular Pathogenesis of ALS
Dr. Randal Tibbetts studies genome surveillance, cell growth and neurodegeneration. Current work in his lab is aimed at understanding how diverse growth-inhibitory signals, including DNA damage, cell-cell contact and growth factor depletion downregulate gene expression through the CREB pathway and how defects in this regulation contribute to tumorigenesis.
Tibbetts Lab
The Tibbetts Lab studies growth control and DNA repair pathways that become disrupted in cancer cells while maintaining secondary interests in neurodegenerative disease.
DNA repair defects and increased proliferative potential are two hallmarks of virtually all cancer cells. We are investigating pathways controlling DNA repair and cell proliferation—and the interfaces between them—to define cancer cell vulnerabilities that may be exploited through novel targeted therapies.
Genome surveillance and cell growth regulation
There are currently two active projects that broadly pertain to mechanisms of genome surveillance growth regulation and tumor suppression.
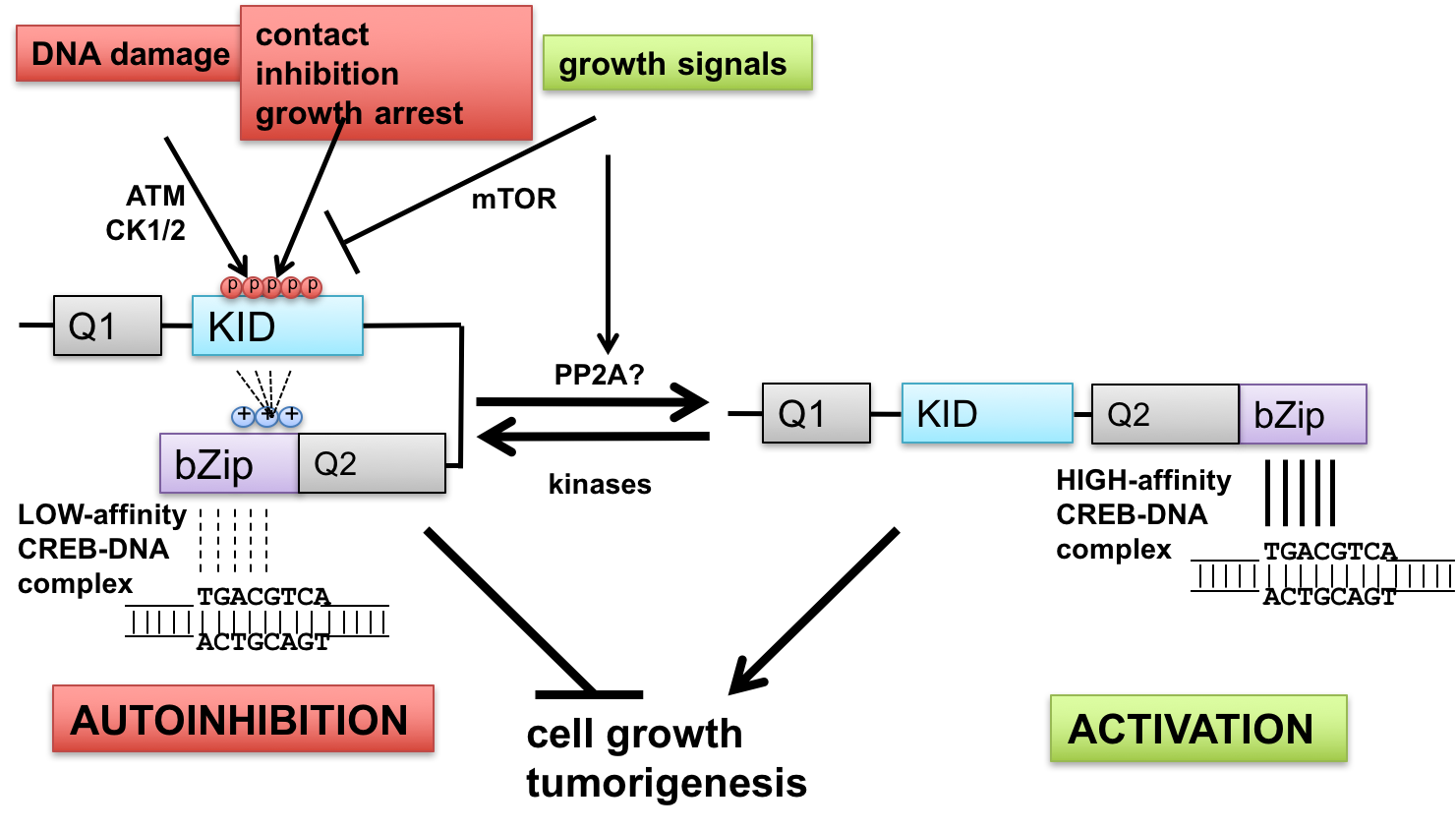
- Interface between DNA damage signaling and CREB-mediated transcription: implications for tumor suppression and metabolic control—The ataxia-telangiectasia-mutated (ATM) protein kinase and related proteins ATR and DNA-PK function as master regulators of the cellular DNA damage response, phosphorylating hundreds of proteins in response to various types of genotoxic stress. Mutations in ATM cause ataxia-telangiectasia (A-T), a syndrome of cancer susceptibility and neurodegeneration characterized at the cellular level by radiation sensitivity and dramatically impaired ability to signal and repair DNA double-strand breaks. A-T patients and ATM-deficient mice also manifest metabolic abnormalities suggesting that ATM senses and responds to metabolic cues. We are investigating how ATM signaling through the CREB (cAMP-response element-binding protein) contributes to cell growth and metabolic control. CREB fulfills key roles in metabolism (gluconeogenesis) and long-term memory formation and has been strongly implicated as a protooncogene in a host of cancers. We have defined a new mechanism of CREB regulation whereby ATM-dependent phosphorylation of a conserved cluster of Ser/Thr residues diminishes CREB DNA binding activity through an autoinhibitory mechanism. In addition, ATM-independent pathways mediate CREB autoinhibition in response to a variety of growth-inhibitory stimuli, whereas signaling through the pro-growth mTOR pathway promotes CREB dephosphorylation (Fig. 1). In addition to defining these pathways and elucidating biochemical mechanisms of autoinhibition, we are currently investigating functional impacts of cancer-associated mutations that may diminish CREB autoinhibition. The importance of these pathways in tumor suppression and other CREB-mediated processes is being tested using CREB gene-edited mice.
- Novel roles of RNA-binding proteins in DNA damage repair and tumor suppression—A second project is focusing on emergent roles for RNA-bindings proteins (RBPs) in DNA damage-dependent alternative splicing, DNA repair and tumor suppression. We are elucidating pathways controlling DNA damage-dependent changes in alternative splicing and modeling the implications of these changes in mice using CRISPR/CAS9-mediated genome editing in mice. The long-term goal of these studies is to understand fundamental aspects of DNA damage repair and response that can be used to guide radio- and chemotherapeutic strategies for cancer patients.
Molecular pathogenesis of ALS.
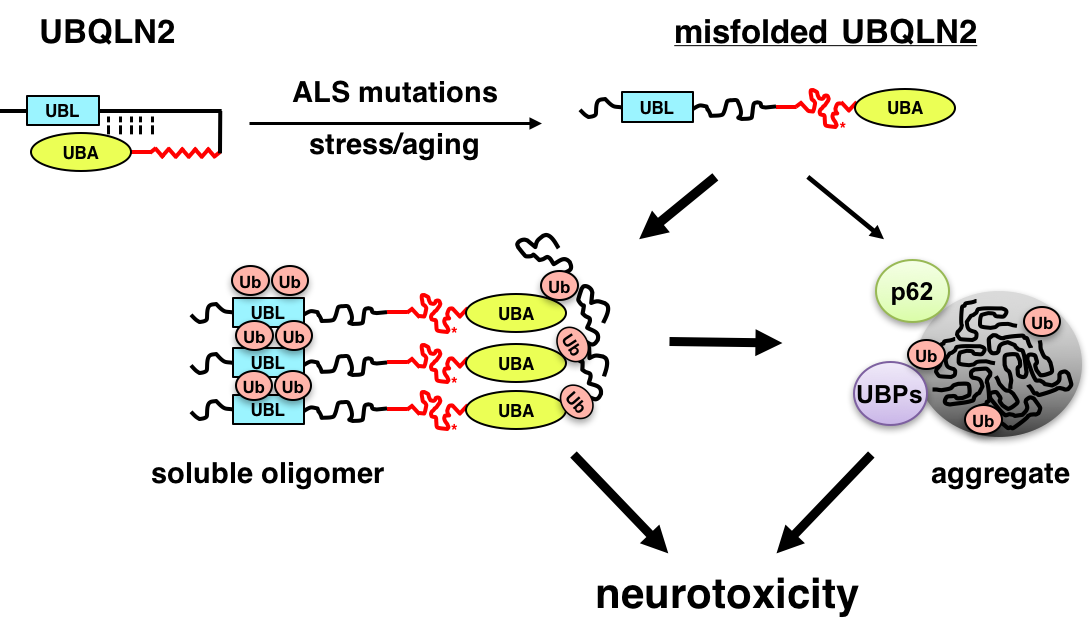
ALS is a fatal neurodegenerative disease that affects motor neurons. Recent genetic advances have identified genes that are critically involved in the ALS disease process, including the RNA-binding proteins TDP-43 and FUS/TLS, the uncharacterized open reading frame, C9ORF72, and UBQLN2. We are using cell culture, Drosophila melanogaster (fruit fly) and mouse models to understand how ALS-associated mutations in these genes instigate neurodegeneration. We are particularly interested in how ALS mutations in the ubiquitin chaperone UBQLN2 promote its misfolding and aggregation, leading to disruption of protein clearance mechanisms and neuron death (Fig. 2). The long-term goal of this work is to identify key pathways that may be amenable to therapeutic intervention in ALS and related dementias.
-
Alternative splicing modulates chromatin interactome and phase separation of the RIF1 C-terminal domain bioRxiv : the preprint server for biology
Koo AS, Jia W, Kim SH, Scalf M, Boos CE, Chen Y, Wang D, Voter AF, Bajaj A, Smith LM, Keck JL, Bakkenist CJ, Guo L, Tibbetts RS
2024 Nov 1:2024.10.29.619708. doi: 10.1101/2024.10.29.619708.
-
More
RIF1 (RAP1 interacting factor) fulfills diverse roles in DNA double-strand break repair, DNA replication, and nuclear organization. RIF1 is expressed as two splice variants, RIF1-Long (RIF1-L) and RIF1-Short (RIF1-S), from the alternative splicing (AS) of Exon 32 (Ex32) which encodes a 26 aa Ser/Lys-rich cassette peptide in the RIF1 C-terminal domain (CTD). Here we demonstrate that Ex32 inclusion was repressed by DNA damage and oncogenesis but peaked at G2/M phase of the cell cycle. Ex32 splice-in was catalyzed by positive regulators including SRSF1, which bound to Ex32 directly, and negative regulators such as PTBP1 and SRSF3. Isoform proteomics revealed enhanced association of RIF1-L with MDC1, whose recruitment to IR-induced foci was strengthened by RIF1-L. RIF1-L and RIF1-S also exhibited unique phase separation and chromatin-binding characteristics that were regulated by CDK1-dependent CTD phosphorylation. These combined findings suggest that regulated AS affects multiple aspects of RIF1 function in genome protection and organization.
PMID:39553946 | PMC:PMC11565852 | DOI:10.1101/2024.10.29.619708
View details for PubMedID 39553946
-
More
-
Axon guidance genes modulate neurotoxicity of ALS-associated UBQLN2 eLife
Kim SH, Nichols KD, Anderson EN, Liu Y, Ramesh N, Jia W, Kuerbis CJ, Scalf M, Smith LM, Pandey UB, Tibbetts RS
2023 Apr 11;12:e84382. doi: 10.7554/eLife.84382.
-
More
Mutations in the ubiquitin (Ub) chaperone Ubiquilin 2 (UBQLN2) cause X-linked forms of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) through unknown mechanisms. Here, we show that aggregation-prone, ALS-associated mutants of UBQLN2 (UBQLN2ALS) trigger heat stress-dependent neurodegeneration in Drosophila. A genetic modifier screen implicated endolysosomal and axon guidance genes, including the netrin receptor, Unc-5, as key modulators of UBQLN2 toxicity. Reduced gene dosage of Unc-5 or its coreceptor Dcc/frazzled diminished neurodegenerative phenotypes, including motor dysfunction, neuromuscular junction defects, and shortened lifespan, in flies expressing UBQLN2ALS alleles. Induced pluripotent stem cells (iPSCs) harboring UBQLN2ALS knockin mutations exhibited lysosomal defects while inducible motor neurons (iMNs) expressing UBQLN2ALS alleles exhibited cytosolic UBQLN2 inclusions, reduced neurite complexity, and growth cone defects that were partially reversed by silencing of UNC5B and DCC. The combined findings suggest that altered growth cone dynamics are a conserved pathomechanism in UBQLN2-associated ALS/FTD.
PMID:37039476 | PMC:PMC10147378 | DOI:10.7554/eLife.84382
View details for PubMedID 37039476
-
More
-
Fused in sarcoma regulates DNA replication timing and kinetics The Journal of biological chemistry
Jia W, Kim SH, Scalf MA, Tonzi P, Millikin RJ, Guns WM, Liu L, Mastrocola AS, Smith LM, Huang TT, Tibbetts RS
2021 Sep;297(3):101049. doi: 10.1016/j.jbc.2021.101049. Epub 2021 Aug 8.
-
More
Fused in sarcoma (FUS) encodes an RNA-binding protein with diverse roles in transcriptional activation and RNA splicing. While oncogenic fusions of FUS and transcription factor DNA-binding domains are associated with soft tissue sarcomas, dominant mutations in FUS can cause amyotrophic lateral sclerosis. FUS has also been implicated in genome maintenance. However, the underlying mechanisms of its actions in genome stability are unknown. Here, we applied gene editing, functional reconstitution, and integrated proteomics and transcriptomics to illuminate roles for FUS in DNA replication and repair. Consistent with a supportive role in DNA double-strand break repair, FUS-deficient cells exhibited subtle alterations in the recruitment and retention of double-strand break-associated factors, including 53BP1 and BRCA1. FUS-/- cells also exhibited reduced proliferative potential that correlated with reduced speed of replication fork progression, diminished loading of prereplication complexes, enhanced micronucleus formation, and attenuated expression and splicing of S-phase-associated genes. Finally, FUS-deficient cells exhibited genome-wide alterations in DNA replication timing that were reversed upon re-expression of FUS complementary DNA. We also showed that FUS-dependent replication domains were enriched in transcriptionally active chromatin and that FUS was required for the timely replication of transcriptionally active DNA. These findings suggest that alterations in DNA replication kinetics and programming contribute to genome instability and functional defects in FUS-deficient cells.
PMID:34375640 | PMC:PMC8403768 | DOI:10.1016/j.jbc.2021.101049
View details for PubMedID 34375640
-
More
-
Roles of constitutive and signal-dependent protein phosphatase 2A docking motifs in burst attenuation of the cyclic AMP response element-binding protein The Journal of biological chemistry
Kim SH, Wu C, Jia W, Xing Y, Tibbetts RS
2021 Jul;297(1):100908. doi: 10.1016/j.jbc.2021.100908. Epub 2021 Jun 24.
-
More
The cAMP response element-binding protein (CREB) is an important regulator of cell growth, metabolism, and synaptic plasticity. CREB is activated through phosphorylation of an evolutionarily conserved Ser residue (S133) within its intrinsically disordered kinase-inducible domain (KID). Phosphorylation of S133 in response to cAMP, Ca2+, and other stimuli triggers an association of the KID with the KID-interacting (KIX) domain of the CREB-binding protein (CBP), a histone acetyl transferase (HAT) that promotes transcriptional activation. Here we addressed the mechanisms of CREB attenuation following bursts in CREB phosphorylation. We show that phosphorylation of S133 is reversed by protein phosphatase 2A (PP2A), which is recruited to CREB through its B56 regulatory subunits. We found that a B56-binding site located at the carboxyl-terminal boundary of the KID (BS2) mediates high-affinity B56 binding, while a second binding site (BS1) located near the amino terminus of the KID mediates low affinity binding enhanced by phosphorylation of adjacent casein kinase (CK) phosphosites. Mutations that diminished B56 binding to BS2 elevated both basal and stimulus-induced phosphorylation of S133, increased CBP interaction with CREB, and potentiated the expression of CREB-dependent reporter genes. Cells from mice harboring a homozygous CrebE153D mutation that disrupts BS2 exhibited increased S133 phosphorylation stoichiometry and elevated transcriptional bursts to cAMP. These findings provide insights into substrate targeting by PP2A holoenzymes and establish a new mechanism of CREB attenuation that has implications for understanding CREB signaling in cell growth, metabolism, synaptic plasticity, and other physiologic contexts.
PMID:34171357 | PMC:PMC8294589 | DOI:10.1016/j.jbc.2021.100908
View details for PubMedID 34171357
-
More
-
Mutation-dependent aggregation and toxicity in a Drosophila model for UBQLN2-associated ALS Human molecular genetics
Kim SH, Stiles SG, Feichtmeier JM, Ramesh N, Zhan L, Scalf MA, Smith LM, Pandey UB, Tibbetts RS
2018 Jan 15;27(2):322-337. doi: 10.1093/hmg/ddx403.
-
More
Members of the conserved ubiquilin (UBQLN) family of ubiquitin (Ub) chaperones harbor an antipodal UBL (Ub-like)-UBA (Ub-associated) domain arrangement and participate in proteasome and autophagosome-mediated protein degradation. Mutations in a proline-rich-repeat region (PRR) of UBQLN2 cause amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD); however, neither the normal functions of the PRR nor impacts of ALS-associated mutations within it are well understood. In this study, we show that ALS mutations perturb UBQLN2 solubility and folding in a mutation-specific manner. Biochemical impacts of ALS mutations were additive, transferable to UBQLN1, and resulted in enhanced Ub association. A Drosophila melanogaster model for UBQLN2-associated ALS revealed that both wild-type and ALS-mutant UBQLN2 alleles disrupted Ub homeostasis; however, UBQLN2ALS mutants exhibited age-dependent aggregation and caused toxicity phenotypes beyond those seen for wild-type UBQLN2. Although UBQLN2 toxicity was not correlated with aggregation in the compound eye, aggregation-prone UBQLN2 mutants elicited climbing defects and neuromuscular junctions (NMJ) abnormalities when expressed in neurons. An UBA domain mutation that abolished Ub binding also diminished UBQLN2 toxicity, implicating Ub binding in the underlying pathomechanism. We propose that ALS-associated mutations in UBQLN2 disrupt folding and that both aggregated species and soluble oligomers instigate neuron autonomous toxicity through interference with Ub homeostasis.
PMID:29161404 | PMC:PMC5886165 | DOI:10.1093/hmg/ddx403
View details for PubMedID 29161404
-
More
-
Tunable regulation of CREB DNA binding activity couples genotoxic stress response and metabolism Nucleic acids research
Kim SH, Trinh AT, Larsen MC, Mastrocola AS, Jefcoate CR, Bushel PR, Tibbetts RS
2016 Nov 16;44(20):9667-9680. doi: 10.1093/nar/gkw643. Epub 2016 Jul 18.
-
More
cAMP response element binding protein (CREB) is a key regulator of glucose metabolism and synaptic plasticity that is canonically regulated through recruitment of transcriptional coactivators. Here we show that phosphorylation of CREB on a conserved cluster of Ser residues (the ATM/CK cluster) by the DNA damage-activated protein kinase ataxia-telangiectasia-mutated (ATM) and casein kinase1 (CK1) and casein kinase2 (CK2) positively and negatively regulates CREB-mediated transcription in a signal dependent manner. In response to genotoxic stress, phosphorylation of the ATM/CK cluster inhibited CREB-mediated gene expression, DNA binding activity and chromatin occupancy proportional to the number of modified Ser residues. Paradoxically, substoichiometric, ATM-independent, phosphorylation of the ATM/CK cluster potentiated bursts in CREB-mediated transcription by promoting recruitment of the CREB coactivator, cAMP-regulated transcriptional coactivators (CRTC2). Livers from mice expressing a non-phosphorylatable CREB allele failed to attenuate gluconeogenic genes in response to DNA damage or fully activate the same genes in response to glucagon. We propose that phosphorylation-dependent regulation of DNA binding activity evolved as a tunable mechanism to control CREB transcriptional output and promote metabolic homeostasis in response to rapidly changing environmental conditions.
PMID:27431323 | PMC:PMC5175338 | DOI:10.1093/nar/gkw643
View details for PubMedID 27431323
-
More
-
Neurodegeneration: Problems at the nuclear pore Nature
Fox BW, Tibbetts RS
2015 Sep 3;525(7567):36-7. doi: 10.1038/nature15208. Epub 2015 Aug 26.
-
Opposing roles of p38 and JNK in a Drosophila model of TDP-43 proteinopathy reveal oxidative stress and innate immunity as pathogenic components of neurodegeneration Human molecular genetics
Zhan L, Xie Q, Tibbetts RS
2015 Feb 1;24(3):757-72. doi: 10.1093/hmg/ddu493. Epub 2014 Oct 3.
-
More
Pathological aggregation and mutation of the 43-kDa TAR DNA-binding protein (TDP-43) are strongly implicated in the pathogenesis amyotrophic lateral sclerosis and frontotemporal lobar degeneration. TDP-43 neurotoxicity has been extensively modeled in mice, zebrafish, Caenorhabditis elegans and Drosophila, where selective expression of TDP-43 in motoneurons led to paralysis and premature lethality. Through a genetic screen aimed to identify genetic modifiers of TDP-43, we found that the Drosophila dual leucine kinase Wallenda (Wnd) and its downstream kinases JNK and p38 influenced TDP-43 neurotoxicity. Reducing Wnd gene dosage or overexpressing its antagonist highwire partially rescued TDP-43-associated premature lethality. Downstream of Wnd, the JNK and p38 kinases played opposing roles in TDP-43-associated neurodegeneration. LOF alleles of the p38b gene as well as p38 inhibitors diminished TDP-43-associated premature lethality, whereas p38b GOF caused phenotypic worsening. In stark contrast, disruptive alleles of Basket (Bsk), the Drosophila homologue of JNK, exacerbated longevity shortening, whereas overexpression of Bsk extended lifespan. Among possible mechanisms, we found motoneuron-directed expression of TDP-43 elicited oxidative stress and innate immune gene activation that were exacerbated by p38 GOF and Bsk LOF, respectively. A key pathologic role for innate immunity in TDP-43-associated neurodegeneration was further supported by the finding that genetic suppression of the Toll/Dif and Imd/Relish inflammatory pathways dramatically extended lifespan of TDP-43 transgenic flies. We propose that oxidative stress and neuroinflammation are intrinsic components of TDP-43-associated neurodegeneration and that the balance between cytoprotective JNK and cytotoxic p38 signaling dictates phenotypic outcome to TDP-43 expression in Drosophila.
PMID:25281658 | PMC:PMC4291250 | DOI:10.1093/hmg/ddu493
View details for PubMedID 25281658
-
More
-
The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage The Journal of biological chemistry
Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS
2013 Aug 23;288(34):24731-41. doi: 10.1074/jbc.M113.497974. Epub 2013 Jul 5.
-
More
The list of factors that participate in the DNA damage response to maintain genomic stability has expanded significantly to include a role for proteins involved in RNA processing. Here, we provide evidence that the RNA-binding protein fused in sarcoma/translocated in liposarcoma (FUS) is a novel component of the DNA damage response. We demonstrate that FUS is rapidly recruited to sites of laser-induced DNA double-strand breaks (DSBs) in a manner that requires poly(ADP-ribose) (PAR) polymerase activity, but is independent of ataxia-telangiectasia mutated kinase function. FUS recruitment is mediated by the arginine/glycine-rich domains, which interact directly with PAR. In addition, we identify a role for the prion-like domain in promoting accumulation of FUS at sites of DNA damage. Finally, depletion of FUS diminished DSB repair through both homologous recombination and nonhomologous end-joining, implicating FUS as an upstream participant in both pathways. These results identify FUS as a new factor in the immediate response to DSBs that functions downstream of PAR polymerase to preserve genomic integrity.
PMID:23833192 | PMC:PMC3750169 | DOI:10.1074/jbc.M113.497974
View details for PubMedID 23833192
-
More
-
Cyclin-dependent kinase 1-dependent phosphorylation of cAMP response element-binding protein decreases chromatin occupancy The Journal of biological chemistry
Trinh AT, Kim SH, Chang H, Mastrocola AS, Tibbetts RS
2013 Aug 16;288(33):23765-75. doi: 10.1074/jbc.M113.464057. Epub 2013 Jun 27.
-
More
The cyclic AMP response element-binding protein (CREB) initiates transcriptional responses to a wide variety of stimuli. CREB activation involves its phosphorylation on Ser-133, which promotes interaction between the CREB kinase-inducible domain (KID) and the KID-interacting domain of the transcriptional coactivator, CREB-binding protein (CBP). The KID also contains a highly conserved phosphorylation cluster, termed the ATM/CK cluster, which is processively phosphorylated in response to DNA damage by the coordinated actions of ataxia-telangiectasia-mutated (ATM) and casein kinases (CKs) 1 and 2. The ATM/CK cluster phosphorylation attenuates CBP binding and CREB transcriptional activity. Paradoxically, it was recently reported that DNA damage activates CREB through homeodomain-interacting protein kinase 2-dependent phosphorylation of Ser-271 near the CREB bZIP DNA binding domain. In this study we sought to further clarify DNA damage-dependent CREB phosphorylation as well as to explore the possibility that the ATM/CK cluster and Ser-271 synergistically or antagonistically modulate CREB activity. We show that, rather than being induced by DNA damage, Ser-270 and Ser-271 of CREB cophosphorylated in a CDK1-dependent manner during G2/M phase. Functionally, we show that phosphorylation of CREB on Ser-270/Ser-271 during mitosis correlated with reduced CREB chromatin occupancy. Furthermore, CDK1-dependent phosphorylation of CREB in vitro inhibited its DNA binding activity. The combined results suggest that CDK1-dependent phosphorylation of CREB on Ser-270/Ser-271 facilitates its dissociation from chromatin during mitosis by reducing its intrinsic DNA binding potential.
PMID:23814058 | PMC:PMC3745323 | DOI:10.1074/jbc.M113.464057
View details for PubMedID 23814058
-
More
-
Identification of genetic modifiers of TDP-43 neurotoxicity in Drosophila PloS one
Zhan L, Hanson KA, Kim SH, Tare A, Tibbetts RS
2013;8(2):e57214. doi: 10.1371/journal.pone.0057214. Epub 2013 Feb 27.
-
More
Cytosolic aggregation of the nuclear RNA-binding protein TDP-43 is a histopathologic signature of degenerating neurons in amyotrophic lateral sclerosis (ALS), and mutations in the TARDBP gene encoding TDP-43 cause dominantly inherited forms of this condition. To understand the relationship between TDP-43 misregulation and neurotoxicity, we and others have used Drosophila as a model system, in which overexpression of either wild-type TDP-43 or its ALS-associated mutants in neurons is sufficient to induce neurotoxicity, paralysis, and early death. Using microarrays, we have examined gene expression patterns that accompany TDP-43-induced neurotoxicity in the fly system. Constitutive expression of TDP-43 in the Drosophila compound eye elicited widespread gene expression changes, with strong upregulation of cell cycle regulatory genes and genes functioning in the Notch intercellular communication pathway. Inducible expression of TDP-43 specifically in neurons elicited significant expression differences in a more restricted set of genes. Genes that were upregulated in both paradigms included SpindleB and the Notch target Hey, which appeared to be a direct TDP-43 target. Mutations that diminished activity of Notch or disrupted the function of downstream Notch target genes extended the lifespan of TDP-43 transgenic flies, suggesting that Notch activation was deleterious in this model. Finally, we showed that mutation of the nucleoporin Nup50 increased the lifespan of TDP-43 transgenic flies, suggesting that nuclear events contribute to TDP-43-dependent neurotoxicity. The combined findings identified pathways whose deregulation might contribute to TDP-43-induced neurotoxicity in Drosophila.
PMID:23468938 | PMC:PMC3584124 | DOI:10.1371/journal.pone.0057214
View details for PubMedID 23468938
-
More
-
High-content RNAi screening identifies the Type 1 inositol triphosphate receptor as a modifier of TDP-43 localization and neurotoxicity Human molecular genetics
Kim SH, Zhan L, Hanson KA, Tibbetts RS
2012 Nov 15;21(22):4845-56. doi: 10.1093/hmg/dds321. Epub 2012 Aug 7.
-
More
Cytosolic aggregation of the nuclear RNA-binding protein (RBP) TDP-43 (43 kDa TAR DNA-binding domain protein) is a suspected direct or indirect cause of motor neuron deterioration in amyotrophic lateral sclerosis (ALS). In this study, we implemented a high-content, genome-wide RNAi screen to identify pathways controlling TDP-43 nucleocytoplasmic shuttling. We identified ∼60 genes whose silencing increased the cytosolic localization of TDP-43, including nuclear pore complex components and regulators of G2/M cell cycle transition. In addition, we identified the type 1 inositol-1,4,5-trisphosphate (IP3) receptor (ITPR1), an IP3-gated, endoplasmic reticulum (ER)-resident Ca(2+) channel, as a strong modulator of TDP-43 nucleocytoplasmic shuttling. Knockdown or chemical inhibition of ITPR1 induced TDP-43 nuclear export in immortalized cells and primary neurons and strongly potentiated the recruitment of TDP-43 to Ubiquilin-positive autophagosomes, suggesting that diminished ITPR1 function leads to autophagosomal clearance of TDP-43. The functional significance of the TDP-43-ITPR1 genetic interaction was tested in Drosophila, where mutant alleles of ITPR1 were found to significantly extended lifespan and mobility of flies expressing TDP-43 under a motor neuron driver. These combined findings implicate IP3-gated Ca(2+) as a key regulator of TDP-43 nucleoplasmic shuttling and proteostasis and suggest pharmacologic inhibition of ITPR1 as a strategy to combat TDP-43-induced neurodegeneration in vivo.
PMID:22872699 | PMC:PMC3529575 | DOI:10.1093/hmg/dds321
View details for PubMedID 22872699
-
More
-
RNA-binding proteins in neurodegenerative disease: TDP-43 and beyond Wiley interdisciplinary reviews. RNA
Hanson KA, Kim SH, Tibbetts RS
2012 Mar-Apr;3(2):265-85. doi: 10.1002/wrna.111. Epub 2011 Oct 25.
-
More
Neurodegenerative diseases are a diverse group of disorders that affect different neuron populations, differ in onset and severity, and can be either inherited or sporadic. One common pathological feature of most of these diseases is the presence of insoluble inclusions in and around neurons, which largely consist of misfolded and aggregated protein. For this reason, neurodegenerative diseases are typically thought to be disorders of aberrant protein processing, in which the cumulative effects of misfolded protein aggregates overwhelm the neuron's proteostatic capacity. However, a growing body of evidence suggests a role for abnormal RNA processing in neurodegenerative disease. The importance of RNA metabolism in disease was highlighted by the discovery of TDP-43 (TAR DNA-binding protein of 43 kDa), an RNA-binding protein (RBP), as a primary component of insoluble aggregates in patients with sporadic amyotrophic lateral sclerosis (ALS). Subsequently, inherited mutations in TDP-43 and the structurally related RBP, FUS/TLS (fused in sarcoma/translated in liposarcoma), were found to cause ALS. These exciting findings have ushered in a new era of ALS research in which the deregulation of RNA metabolism is viewed as a central cause of motor neuron deterioration. In addition, the fact that neuropathologically and anatomically distinct neurodegenerative diseases display altered RNA metabolism suggests that common pathologic mechanisms may underlie many of these disorders.
PMID:22028183 | PMC:PMC3766724 | DOI:10.1002/wrna.111
View details for PubMedID 22028183
-
More
-
Casein kinase 1-dependent phosphorylation of familial advanced sleep phase syndrome-associated residues controls PERIOD 2 stability The Journal of biological chemistry
Shanware NP, Hutchinson JA, Kim SH, Zhan L, Bowler MJ, Tibbetts RS
2011 Apr 8;286(14):12766-74. doi: 10.1074/jbc.M111.224014. Epub 2011 Feb 15.
-
More
The mammalian circadian clock component PERIOD2 (PER2) plays a critical role in circadian rhythm entrainment. Recently, a missense mutation at a putative phosphorylation site in hPER2, Ser-662, was identified in patients that suffer from familial advanced sleep phase syndrome (FASPS). Patients with FASPS display abnormal sleep-wake patterns characterized by a lifelong pattern of sleep onset in the early evening and offset in the early morning. Although the phosphorylation of PER2 is strongly implied from functional studies, it has not been possible to study the site-specific phosphorylation of PER2 on Ser-662, and the biochemical functions of this residue are unclear. Here, we used phospho-specific antibodies to show that PER2 is phosphorylated on Ser-662 and flanking casein kinase (CK) sites in vivo. The phosphorylation of PER2 was carried out by the combined activities of casein kinase 1δ (CK1 δ) and casein kinase 1ε (CK1ε) and was antagonized by protein phosphatase 1. PER2 phosphorylation was rapidly induced in response to circadian entrainment of mammalian cell lines and occurred in both cytosolic and nuclear compartments. Importantly, we found that the pool of Ser-662-phosphorylated PER2 proteins was more stable than the pool of total PER2 molecules, implying that the FASPS phosphorylation cluster antagonizes PER2 degradation. Consistent with this idea, a Ser-662→Ala mutation that abrogated PER2 phosphorylation significantly reduced its half-life, whereas a phosphomimetic Ser-662→Asp substitution led to an elevation in half-life. Our combined findings provide new insights into PER2 regulation and the biochemical basis of FASPS.
PMID:21324900 | PMC:PMC3069476 | DOI:10.1074/jbc.M111.224014
View details for PubMedID 21324900
-
More
-
Regulation of ribosomal protein S6 phosphorylation by casein kinase 1 and protein phosphatase 1 The Journal of biological chemistry
Hutchinson JA, Shanware NP, Chang H, Tibbetts RS
2011 Mar 11;286(10):8688-8696. doi: 10.1074/jbc.M110.141754. Epub 2011 Jan 13.
-
More
Ribosomal protein S6 (rpS6) is a critical component of the 40 S ribosomal subunit that mediates translation initiation at the 5'-m(7)GpppG cap of mRNA. In response to mitogenic stimuli, rpS6 undergoes ordered C-terminal phosphorylation by p70 S6 kinases and p90 ribosomal S6 kinases on four conserved Ser residues (Ser-235, Ser-236, Ser-240, and Ser-244) whose modification potentiates rpS6 cap binding activity. A fifth site, Ser-247, is also known to be phosphorylated, but its function and regulation are not well characterized. In this study, we employed phospho-specific antibodies to show that Ser-247 is a target of the casein kinase 1 (CK1) family of protein kinases. CK1-dependent phosphorylation of Ser-247 was induced by mitogenic stimuli and required prior phosphorylation of upstream S6 kinase/ribosomal S6 kinase residues. CK1-mediated phosphorylation of Ser-247 also enhanced the phosphorylation of upstream sites, which implies that bidirectional synergy between C-terminal phospho-residues is required to sustain rpS6 phosphorylation. Consistent with this idea, CK1-dependent phosphorylation of rpS6 promotes its association with the mRNA cap-binding complex in vitro. Additionally, we show that protein phosphatase 1 (PP1) antagonizes rpS6 C terminus phosphorylation and cap binding in intact cells. These findings further our understanding of rpS6 phospho-regulation and define a direct link between CK1 and translation initiation.
PMID:21233202 | PMC:PMC3048750 | DOI:10.1074/jbc.M110.141754
View details for PubMedID 21233202
-
More
-
Conserved and distinct modes of CREB/ATF transcription factor regulation by PP2A/B56gamma and genotoxic stress PloS one
Shanware NP, Zhan L, Hutchinson JA, Kim SH, Williams LM, Tibbetts RS
2010 Aug 13;5(8):e12173. doi: 10.1371/journal.pone.0012173.
-
More
Activating transcription factor 1 (ATF1) and the closely related proteins CREB (cyclic AMP resonse element binding protein) and CREM (cyclic AMP response element modulator) constitute a subfamily of bZIP transcription factors that play critical roles in the regulation of cellular growth, metabolism, and survival. Previous studies demonstrated that CREB is phosphorylated on a cluster of conserved Ser residues, including Ser-111 and Ser-121, in response to DNA damage through the coordinated actions of the ataxia-telangiectasia-mutated (ATM) protein kinase and casein kinases 1 and 2 (CK1/2). Here, we show that DNA damage-induced phosphorylation by ATM is a general feature of CREB and ATF1. ATF1 harbors a conserved ATM/CK cluster that is constitutively and stoichiometrically phosphorylated by CK1 and CK2 in asynchronously growing cells. Exposure to DNA damage further induced ATF1 phosphorylation on Ser-51 by ATM in a manner that required prior phosphorylation of the upstream CK residues. Hyperphosphorylated ATF1 showed a 4-fold reduced affinity for CREB-binding protein. We further show that PP2A, in conjunction with its targeting subunit B56gamma, antagonized ATM and CK1/2-dependent phosphorylation of CREB and ATF1 in cellulo. Finally, we show that CK sites in CREB are phosphorylated during cellular growth and that phosphorylation of these residues reduces the threshold of DNA damage required for ATM-dependent phosphorylation of the inhibitory Ser-121 residue. These studies define overlapping and distinct modes of CREB and ATF1 regulation by phosphorylation that may ensure concerted changes in gene expression mediated by these factors.
PMID:20730097 | PMC:PMC2921338 | DOI:10.1371/journal.pone.0012173
View details for PubMedID 20730097
-
More
-
Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA The Journal of biological chemistry
Kim SH, Shanware NP, Bowler MJ, Tibbetts RS
2010 Oct 29;285(44):34097-105. doi: 10.1074/jbc.M110.154831. Epub 2010 Aug 18.
-
More
Amyotrophic lateral sclerosis (ALS) is an incurable neurodegenerative disease that preferentially targets motor neurons. It was recently found that dominant mutations in two related RNA-binding proteins, TDP-43 (43-kDa TAR DNA-binding domain protein) and FUS/TLS (fused in sarcoma/translated in liposarcoma) cause a subset of ALS. The convergent ALS phenotypes associated with TDP-43 and FUS/TLS mutations are suggestive of a functional relationship; however, whether or not TDP-43 and FUS/TLS operate in common biochemical pathways is not known. Here we show that TDP-43 and FUS/TLS directly interact to form a complex at endogenous expression levels in mammalian cells. Binding was mediated by an unstructured TDP-43 C-terminal domain and occurred within the context of a 300-400-kDa complex that also contained C-terminal cleavage products of TDP-43 linked to neuropathology. TDP-43 C-terminal fragments were excluded from large molecular mass TDP-43 ribonucleoprotein complexes but retained FUS/TLS binding activity. The functional significance of TDP-43-FUS/TLS complexes was established by showing that RNAi silencing of either TDP-43 or FUS/TLS reduced the expression of histone deacetylase (HDAC) 6 mRNA. TDP-43 and FUS/TLS associated with HDAC6 mRNA in intact cells and in vitro, and competition experiments suggested that the proteins occupy overlapping binding sites. The combined findings demonstrate that TDP-43 and FUS/TLS form a functional complex in intact cells and suggest that convergent ALS phenotypes associated with TDP-43 and FUS/TLS mutations may reflect their participation in common biochemical processes.
PMID:20720006 | PMC:PMC2962508 | DOI:10.1074/jbc.M110.154831
View details for PubMedID 20720006
-
More
-
Transcription-dependent activation of ataxia telangiectasia mutated prevents DNA-dependent protein kinase-mediated cell death in response to topoisomerase I poison The Journal of biological chemistry
Sakasai R, Teraoka H, Takagi M, Tibbetts RS
2010 May 14;285(20):15201-15208. doi: 10.1074/jbc.M110.101808. Epub 2010 Mar 19.
-
More
Camptothecin (CPT) is a topoisomerase I inhibitor, derivatives of which are being used for cancer chemotherapy. CPT-induced DNA double-strand breaks (DSBs) are considered a major cause of its tumoricidal activity, and it has been shown that CPT induces DNA damage signaling through the phosphatidylinositol 3-kinase-related kinases, including ATM (ataxia telangiectasia mutated), ATR (ATM and Rad3-related), and DNA-PK (DNA-dependent protein kinase). In addition, CPT causes DNA strand breaks mediated by transcription, although the downstream signaling events are less well characterized. In this study, we show that CPT-induced activation of ATM requires transcription. Mechanistically, transcription inhibition suppressed CPT-dependent activation of ATM and blocked recruitment of the DNA damage mediator p53-binding protein 1 (53BP1) to DNA damage sites, whereas ATM inhibition abrogated CPT-induced G(1)/S and S phase checkpoints. Functional inactivation of ATM resulted in DNA replication-dependent hyperactivation of DNA-PK in CPT-treated cells and dramatic CPT hypersensitivity. On the other hand, simultaneous inhibition of ATM and DNA-PK partially restored CPT resistance, suggesting that activation of DNA-PK is proapoptotic in the absence of ATM. Correspondingly, comet assay and cell cycle synchronization experiments suggested that transcription collapse occurring as the result of CPT treatment are converted to frank double-strand breaks when ATM-deficient cells bypass the G(1)/S checkpoint. Thus, ATM suppresses DNA-PK-dependent cell death in response to topoisomerase poisons, a finding with potential clinical implications.
PMID:20304914 | PMC:PMC2865312 | DOI:10.1074/jbc.M110.101808
View details for PubMedID 20304914
-
More
-
Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS) The Journal of biological chemistry
Hanson KA, Kim SH, Wassarman DA, Tibbetts RS
2010 Apr 9;285(15):11068-72. doi: 10.1074/jbc.C109.078527. Epub 2010 Feb 12.
-
More
TDP-43 (43-kDa TAR DNA-binding protein) is a major constituent of ubiquitin-positive cytosolic aggregates present in neurons of patients with amyotrophic lateral sclerosis (ALS) and ubiquitin-positive fronto-temporal lobar degeneration (FTLD-U). Inherited mutations in TDP-43 have been linked to familial forms of ALS, indicating a key role for TDP-43 in disease pathogenesis. Here, we describe a Drosophila melanogaster model of TDP-43 proteinopathy. Expression of wild-type human TDP-43 protein in Drosophila motor neurons led to motor dysfunction and dramatic reduction of life span. Interestingly, coexpression of ubiquilin 1, a previously identified TDP-43-interacting protein with suspected functions in autophagy and proteasome targeting, reduced steady-state TDP-43 expression but enhanced the severity of TDP-43 phenotypes. Finally, ectopically expressed TDP-43 was largely localized to motor neuron nuclei, suggesting that expression of wild-type TDP-43 alone is detrimental even in the absence of cytosolic aggregation. Our findings demonstrate that TDP-43 exerts cell-autonomous neurotoxicity in Drosophila and further imply that dose-dependent alterations of TDP-43 nuclear function may underlie motor neuron death in ALS.
PMID:20154090 | PMC:PMC2856981 | DOI:10.1074/jbc.C109.078527
View details for PubMedID 20154090
-
More
-
Proteasome inhibition suppresses DNA-dependent protein kinase activation caused by camptothecin DNA repair
Sakasai R, Teraoka H, Tibbetts RS
2010 Jan 2;9(1):76-82. doi: 10.1016/j.dnarep.2009.10.008. Epub 2009 Dec 3.
-
More
The ubiquitin-proteasome pathway plays an important role in DNA damage signaling and repair by facilitating the recruitment and activation of DNA repair factors and signaling proteins at sites of damaged chromatin. Proteasome activity is generally not thought to be required for activation of apical signaling kinases including the PI3K-related kinases (PIKKs) ATM, ATR, and DNA-PK that orchestrate downstream signaling cascades in response to diverse genotoxic stimuli. In a previous work, we showed that inhibition of the proteasome by MG-132 suppressed 53BP1 (p53 binding protein1) phosphorylation as well as RPA2 (replication protein A2) phosphorylation in response to the topoisomerase I (TopI) poison camptothecin (CPT). To address the mechanism of proteasome-dependent RPA2 phosphorylation, we investigated the effects of proteasome inhibitors on the upstream PIKKs. MG-132 sharply suppressed CPT-induced DNA-PKcs autophosphorylation, a marker of the activation, whereas the phosphorylation of ATM and ATR substrates was only slightly suppressed by MG-132, suggesting that DNA-PK among the PIKKs is specifically regulated by the proteasome in response to CPT. On the other hand, MG-132 did not suppress DNA-PK activation in response to UV or IR. MG-132 blocked the interaction between DNA-PKcs and Ku heterodimer enhanced by CPT, and hydroxyurea pre-treatment completely abolished CPT-induced DNA-PKcs autophosphorylation, indicating a requirement for ongoing DNA replication. CPT-induced TopI degradation occurred independent of DNA-PK activation, suggesting that DNA-PK activation does not require degradation of trapped TopI complexes. The combined results suggest that CPT-dependent replication fork collapse activates DNA-PK signaling through a proteasome dependent, TopI degradation-independent pathway. The implications of DNA-PK activation in the context of TopI poison-based therapies are discussed.
PMID:19959400 | PMC:PMC2818427 | DOI:10.1016/j.dnarep.2009.10.008
View details for PubMedID 19959400
-
More
-
Non-specific in vivo inhibition of CK1 by the pyridinyl imidazole p38 inhibitors SB 203580 and SB 202190 BMB reports
Shanware NP, Williams LM, Bowler MJ, Tibbetts RS
2009 Mar 31;42(3):142-7. doi: 10.5483/bmbrep.2009.42.3.142.
-
More
Small-molecule inhibitors of protein kinases have contributed immensely to our understanding of biological signaling pathways and have been exploited therapeutically for the treatment of cancers and other disease states. The pyridinyl imidazole compounds SB 203580 and SB 202190 were identified as ATP competitive antagonists of the p38 stress-activated protein kinases and have been widely used to elucidate p38-dependent cellular processes. Here, we identify SB 203580 and SB 202190 as potent inhibitors of stress-induced CREB phosphorylation on Serine 111 (Ser-111) in intact cells. Unexpectedly, we found that the inhibitory activity of SB 203580 and SB 202190 on CREB phosphorylation was independent of p38, but instead correlated with inhibition of casein kinase 1 (CK1) in vitro. The inhibition of CK1-mediated CREB phosphorylation by concentrations of pyridinyl imidazoles commonly employed to suppress p38, suggests that in some cases conclusions of p38-dependence derived solely from the use of these inhibitors may be invalid.
PMID:19336000 | PMC:PMC4412876 | DOI:10.5483/bmbrep.2009.42.3.142
View details for PubMedID 19336000
-
More
-
Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the proteasome-targeting factor, ubiquilin 1 The Journal of biological chemistry
Kim SH, Shi Y, Hanson KA, Williams LM, Sakasai R, Bowler MJ, Tibbetts RS
2009 Mar 20;284(12):8083-92. doi: 10.1074/jbc.M808064200. Epub 2008 Dec 26.
-
More
TDP-43 (43-kDa TAR DNA-binding domain protein) is a major constituent of ubiquitin-positive cytoplasmic aggregates present in neurons of patients with fronto-temporal lobular dementia and amyotrophic lateral sclerosis (ALS). The pathologic significance of TDP-43 aggregation is not known; however, dominant mutations in TDP-43 cause a subset of ALS cases, suggesting that misfolding and/or altered trafficking of TDP-43 is relevant to the disease process. Here, we show that the presenilin-binding protein ubiquilin 1 (UBQLN) plays a role in TDP-43 aggregation. TDP-43 interacted with UBQLN both in yeast and in vitro, and the carboxyl-terminal ubiquitin-associated domain of UBQLN was both necessary and sufficient for binding to polyubiquitylated forms of TDP-43. Overexpression of UBQLN recruited TDP-43 to detergent-resistant cytoplasmic aggregates that colocalized with the autophagosomal marker, LC3. UBQLN-dependent aggregation required the UBQLN UBA domain, was mediated by non-overlapping regions of TDP-43, and was abrogated by a mutation in UBQLN previously linked to Alzheimer disease. Four ALS-associated alleles of TDP-43 also coaggregated with UBQLN, and the extent of aggregation correlated with in vitro UBQLN binding affinity. Our findings suggest that UBQLN is a polyubiquitin-TDP-43 cochaperone that mediates the autophagosomal delivery and/or proteasome targeting of TDP-43 aggregates.
PMID:19112176 | PMC:PMC2658102 | DOI:10.1074/jbc.M808064200
View details for PubMedID 19112176
-
More
-
Mutations in String/CDC25 inhibit cell cycle re-entry and neurodegeneration in a Drosophila model of Ataxia telangiectasia Genes & development
Rimkus SA, Katzenberger RJ, Trinh AT, Dodson GE, Tibbetts RS, Wassarman DA
2008 May 1;22(9):1205-20. doi: 10.1101/gad.1639608. Epub 2008 Apr 11.
-
More
Mutations in ATM (Ataxia telangiectasia mutated) result in Ataxia telangiectasia (A-T), a disorder characterized by progressive neurodegeneration. Despite advances in understanding how ATM signals cell cycle arrest, DNA repair, and apoptosis in response to DNA damage, it remains unclear why loss of ATM causes degeneration of post-mitotic neurons and why the neurological phenotype of ATM-null individuals varies in severity. To address these issues, we generated a Drosophila model of A-T. RNAi knockdown of ATM in the eye caused progressive degeneration of adult neurons in the absence of exogenously induced DNA damage. Heterozygous mutations in select genes modified the neurodegeneration phenotype, suggesting that genetic background underlies variable neurodegeneration in A-T. The neuroprotective activity of ATM may be negatively regulated by deacetylation since mutations in a protein deacetylase gene, RPD3, suppressed neurodegeneration, and a human homolog of RPD3, histone deacetylase 2, bound ATM and abrogated ATM activation in cell culture. Moreover, knockdown of ATM in post-mitotic neurons caused cell cycle re-entry, and heterozygous mutations in the cell cycle activator gene String/CDC25 inhibited cell cycle re-entry and neurodegeneration. Thus, we hypothesize that ATM performs a cell cycle checkpoint function to protect post-mitotic neurons from degeneration and that cell cycle re-entry causes neurodegeneration in A-T.
PMID:18408079 | PMC:PMC2335316 | DOI:10.1101/gad.1639608
View details for PubMedID 18408079
-
More
-
Identification of carboxyl-terminal MCM3 phosphorylation sites using polyreactive phosphospecific antibodies The Journal of biological chemistry
Shi Y, Dodson GE, Mukhopadhyay PS, Shanware NP, Trinh AT, Tibbetts RS
2007 Mar 23;282(12):9236-43. doi: 10.1074/jbc.M609256200. Epub 2007 Jan 23.
-
More
The functionally related ATM (ataxia telangiectasia-mutated) and ATR (ATM-Rad3-related) protein kinases are critical regulators of DNA damage responses in mammalian cells. ATM and ATR share highly overlapping substrate specificities and show a strong preference for the phosphorylation of Ser or Thr residues followed by Gln. In this report we used a polyreactive phosphospecific antibody (alpha-pDSQ) that recognizes a subset of phosphorylated Asp-Ser-Gln sequences to purify candidate ATM/ATR substrates. This led to the identification of phosphorylation sites in the carboxyl terminus of the minichromosome maintenance protein 3 (MCM3), a component of the hexameric MCM DNA helicase. We show that the alpha-DSQ antibody recognizes tandem DSQ phosphorylation sites (Ser-725 and Ser-732) in the carboxyl terminus of murine MCM3 (mMCM3) and that ATM phosphorylates both sites in vitro. ATM phosphorylated the carboxyl termini of mMCM3 and human MCM3 in vivo and the phosphorylated form of MCM3 retained association with the canonical MCM complex. Although DNA damage did not affect steady-state levels of chromatin-bound MCM3, the ATM-phosphorylated form of MCM3 was preferentially localized to the soluble, nucleoplasmic fraction. This finding suggests that the carboxyl terminus of chromatin-loaded MCM3 may be sequestered from ATM-dependent checkpoint signals. Finally, we show that ATM and ATR jointly contribute to UV light-induced MCM3 phosphorylation, but that ATM is the predominant UV-activated MCM3 kinase in vivo. The carboxyl-terminal ATM phosphorylation sites are conserved in vertebrate MCM3 orthologs suggesting that this motif may serve important regulatory functions in response to DNA damage. Our findings also suggest that DSQ motifs are common phosphoacceptor motifs for ATM family kinases.
PMID:17244605 | DOI:10.1074/jbc.M609256200
View details for PubMedID 17244605
-
More
-
Coregulated ataxia telangiectasia-mutated and casein kinase sites modulate cAMP-response element-binding protein-coactivator interactions in response to DNA damage The Journal of biological chemistry
Shanware NP, Trinh AT, Williams LM, Tibbetts RS
2007 Mar 2;282(9):6283-91. doi: 10.1074/jbc.M610674200. Epub 2007 Jan 5.
-
More
The cyclic AMP-response element-binding protein (CREB) is a bZIP family transcription factor implicated as an oncoprotein and neuron survival factor. CREB is activated in response to cellular stimuli, including cAMP and Ca(2+), via phosphorylation of Ser-133, which promotes interaction between the kinase-inducible domain (KID) of CREB and the KID-interacting domain of CREB-binding protein (CBP). We previously demonstrated that the interaction between CREB and CBP is inhibited by DNA-damaging stimuli through a mechanism whereby CREB is phosphorylated by the ataxia telangiectasia-mutated (ATM) protein kinase. We now show that the ATM phosphorylation sites in CREB are functionally intertwined with a cluster of coregulated casein kinase (CK) sites. We demonstrate that DNA damage-induced phosphorylation of CREB occurs in three steps. The initial event in the CREB phosphorylation cascade is the phosphorylation of Ser-111, which is carried out by CK1 and CK2 under basal conditions and by ATM in response to ionizing radiation. The phosphorylation of Ser-111 triggers the CK2-dependent phosphorylation of Ser-108 and the CK1-dependent phosphorylation of Ser-114 and Ser-117. The phosphorylation of Ser-114 and Ser-117 by CK1 then renders CREB permissive for ATM-dependent phosphorylation on Ser-121. Mutation of Ser-121 alone abrogates ionizing radiation-dependent repression of CREB-CBP complexes, which can be recapitulated using a CK1 inhibitor. Our findings outline a complex mechanism of CREB phosphorylation in which coregulated ATM and CK sites control CREB transactivation potential by modulating its CBP-binding affinity. The coregulated ATM and CK sites identified in CREB may constitute a signaling motif that is common to other DNA damage-regulated substrates.
PMID:17209043 | DOI:10.1074/jbc.M610674200
View details for PubMedID 17209043
-
More
-
Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli Science (New York, N.Y.)
Wu Z, Shi Y, Tibbetts RS, Miyamoto S
2006 Feb 24;311(5764):1141-6. doi: 10.1126/science.1121513.
-
More
The transcription factor NF-kappaB modulates apoptotic responses induced by genotoxic stress. We show that NF-kappaB essential modulator (NEMO), the regulatory subunit of IkappaB kinase (IKK) (which phosphorylates the NF-kappaB inhibitor IkappaB), associates with activated ataxia telangiectasia mutated (ATM) after the induction of DNA double-strand breaks. ATM phosphorylates serine-85 of NEMO to promote its ubiquitin-dependent nuclear export. ATM is also exported in a NEMO-dependent manner to the cytoplasm, where it associates with and causes the activation of IKK in a manner dependent on another IKK regulator, a protein rich in glutamate, leucine, lysine, and serine (ELKS). Thus, regulated nuclear shuttling of NEMO links two signaling kinases, ATM and IKK, to activate NF-kappaB by genotoxic signals.
PMID:16497931 | DOI:10.1126/science.1121513
View details for PubMedID 16497931
-
More
-
DNA replication stress-induced phosphorylation of cyclic AMP response element-binding protein mediated by ATM The Journal of biological chemistry
Dodson GE, Tibbetts RS
2006 Jan 20;281(3):1692-7. doi: 10.1074/jbc.M509577200. Epub 2005 Nov 17.
-
More
The DNA damage-response regulators ATM (ataxia-telangiectasia-mutated) and ATR (ATM-Rad3-related) are structurally and functionally related protein kinases that exhibit nearly identical substrate specificities in vitro. Current paradigms hold that the relative contributions of ATM and ATR to nuclear substrate phosphorylation are dictated by the type of initiating DNA lesion; ATM-dependent substrate phosphorylation is principally activated by DNA double strand breaks, whereas ATR-dependent substrate phosphorylation is induced by UV light and other forms of DNA replication stress. In this report, we employed the cyclic AMP-response element-binding (CREB) protein to provide evidence for substrate discrimination by ATM and ATR in cellulo. ATM and ATR phosphorylate CREB in vitro, and CREB is phosphorylated on Ser-121 in intact cells in response to ionizing radiation (IR), UV light, and hydroxyurea. The UV light- and hydroxyurea-induced phosphorylation of CREB was delayed in comparison to the canonical ATR substrate CHK1, suggesting potentially different mechanisms of phosphorylation. UV light-induced CREB phosphorylation temporally correlated with ATM autophosphorylation on Ser-1981, and an ATM-specific small interfering RNA suppressed CREB phosphorylation in response to this stimulus. UV light-induced CREB phosphorylation was absent in ATM-deficient cells, confirming that ATM is required for CREB phosphorylation in UV irradiation-damaged cells. Interestingly, RNA interference-mediated suppression of ATR partially inhibited CREB phosphorylation in response to UV light, which correlated with reduced phosphorylation of ATM on Ser-1981. These findings suggest that ATM is the major genotoxin-induced CREB kinase in mammalian cells and that ATR lies upstream of ATM in a UV light-induced signaling pathway.
PMID:16293623 | DOI:10.1074/jbc.M509577200
View details for PubMedID 16293623
-
More
-
ATR activation necessary but not sufficient for p53 induction and apoptosis in hydroxyurea-hypersensitive myeloid leukemia cells Cell cycle (Georgetown, Tex.)
Kumar S, Dodson GE, Trinh A, Puchalski JR, Tibbetts RS
2005 Nov;4(11):1667-74. doi: 10.4161/cc.4.11.2169. Epub 2005 Nov 13.
-
More
Hydroxyurea (HU) is a competitive inhibitor of ribonucleotide reductase that is used for the treatment of myeloproliferative disorders. HU inhibits DNA replication and induces apoptosis in a cell type-dependent manner, yet the relevant pathways that mediate apoptosis in response to this agent are not well characterized. In this study, we employed the human myeloid leukemia 1 (ML-1) cell line as a model to investigate the mechanisms of HU-induced apoptosis. Exposure of ML-1 cells to HU caused rapid cell death that was accompanied by hallmark features of apoptosis, including membrane blebbing, phosphatidylserine translocation, and caspase activation. HU-induced apoptosis required new protein synthesis, was induced by HU exposures as short as 15 min, and correlated with the accumulation of p53 and induction of the p53 target gene PUMA. p53 induction in ML-1 cells was ATR dependent and downregulation of p53 through RNAi delayed HU-induced apoptosis. HU did not induce p53 or induce apoptosis in Molt-3 leukemia cells, even though exposure to HU induced a comparable level of DNA damage and robustly activated the ATR pathway. The microtubule inhibitor nocodazole suppressed HU-induced p53 accumulation in ML-1 cells suggesting that a microtubule-dependent event contributes to p53 induction and apoptosis in this cell line. Our findings outline an HU-induced cell death pathway and suggest that activation of ATR is necessary, but not sufficient, for stabilization of p53 in response to DNA replication stress.
PMID:16258278 | DOI:10.4161/cc.4.11.2169
View details for PubMedID 16258278
-
More
-
Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication in vivo The Journal of biological chemistry
Shi Y, Dodson GE, Shaikh S, Rundell K, Tibbetts RS
2005 Dec 2;280(48):40195-200. doi: 10.1074/jbc.C500400200. Epub 2005 Oct 11.
-
More
The structurally related ATM (ataxia-telangiectasia-mutated) and ATR (ATM-Rad3-related) protein kinases fulfill overlapping yet non-redundant functions as key regulators of cellular DNA damage responses. We recently showed that ATM phosphorylates the cyclic AMP response element-binding protein, CREB, following exposure to ionizing radiation (IR) and other DNA-damaging stimuli. Here, we show that a phospho-specific antibody recognizing the major ATM phosphorylation site in CREB cross-reacts with SV40 large tumor antigen (LTag), a multifunctional oncoprotein required for replication of the SV40 minichromosome. The relevant IR-induced phosphorylation site in LTag recognized by phospho-CREB antibody was mapped to Ser-120. IR strongly induced the phosphorylation of Ser-120 in an ATM-dependent manner in mouse embryo fibroblasts. Infection of African green monkey CV1 cells with SV40 resulted in the activation of ATM and phosphorylation of LTag and endogenous ATM substrates. Infection-induced LTag phosphorylation correlated with the onset of DNA replication, was ATM-dependent, and peaked when viral DNA levels reached their maximum. SV40 replication in CV1 cells required an intact LTag Ser-120 phosphorylation site and was inhibited following transfection with ATM small interfering RNA suggesting that ATM is required for optimal SV40 replication in primate cells. Our findings uncover a direct link between ATM and SV40 LTag that may have implications for understanding the replication cycle of oncogenic polyoma viruses.
PMID:16221684 | DOI:10.1074/jbc.C500400200
View details for PubMedID 16221684
-
More
-
Cell biology. Guiding ATM to broken DNA Science (New York, N.Y.)
Abraham RT, Tibbetts RS
2005 Apr 22;308(5721):510-1. doi: 10.1126/science.1112069.
-
DNA replication defects, spontaneous DNA damage, and ATM-dependent checkpoint activation in replication protein A-deficient cells The Journal of biological chemistry
Dodson GE, Shi Y, Tibbetts RS
2004 Aug 6;279(32):34010-4. doi: 10.1074/jbc.C400242200. Epub 2004 Jun 14.
-
More
Replication protein A (RPA) is a heterotrimeric, single-stranded DNA-binding complex comprised of 70-kDa (RPA1), 32-kDa (RPA2), and 14-kDa (RPA3) subunits that is essential for DNA replication, recombination, and repair in eukaryotes. In addition, recent studies using vertebrate model systems have suggested an important role for RPA in the initiation of cell cycle checkpoints following exposure to DNA replication stress. Specifically, RPA has been implicated in the recruitment and activation of the ATM-Rad3-related protein kinase, ATR, which in conjunction with the related kinase, ATM (ataxia-telangiectasia-mutated), transmits checkpoint signals via the phosphorylation of downstream effectors. In this report, we have explored the effects of RPA insufficiency on DNA replication, cell survival, and ATM/ATR-dependent signal transduction in response to genotoxic stress. RNA interference-mediated suppression of RPA1 caused a slowing of S phase progression, G2/M cell cycle arrest, and apoptosis in HeLa cells. RPA-deficient cells demonstrated high levels of spontaneous DNA damage and constitutive activation of ATM, which was responsible for the terminal G2/M arrest phenotype. Surprisingly, we found that neither RPA1 nor RPA2 were essential for the hydroxyurea- or UV-induced phosphorylation of the ATR substrates CHK1 and CREB (cyclic AMP-response element-binding protein). These findings reveal that RPA is required for genomic stability and suggest that activation of ATR can occur through RPA-independent pathways.
PMID:15197179 | DOI:10.1074/jbc.C400242200
View details for PubMedID 15197179
-
More
-
The mRNA surveillance protein hSMG-1 functions in genotoxic stress response pathways in mammalian cells Molecular cell
Brumbaugh KM, Otterness DM, Geisen C, Oliveira V, Brognard J, Li X, Lejeune F, Tibbetts RS, Maquat LE, Abraham RT
2004 Jun 4;14(5):585-98. doi: 10.1016/j.molcel.2004.05.005.
-
More
Members of the PI 3-kinase-related kinase (PIKK) family function in mitogenic and stress-induced signaling pathways in eukaryotic cells. Here, we characterize the newest PIKK family member, hSMG-1, as a genotoxic stress-activated protein kinase that displays some functional overlap with the related kinase, ATM, in human cells. Both ATM and hSMG-1 phosphorylate Ser/Thr-Gln-containing target sequences in the checkpoint protein p53 and the nonsense-mediated mRNA decay (NMD) protein hUpf1. Expression of hSMG-1 is required for optimal p53 activation after cellular exposure to genotoxic stress, and depletion of hSMG-1 leads to spontaneous DNA damage and increased sensitivity to ionizing radiation (IR). Moreover, IR exposure triggers hUpf1 phosphorylation at Ser/Thr-Gln motifs, and both ATM and hSMG-1 contribute to these phosphorylation events. Finally, NMD is suppressed in hSMG-1- but not ATM-deficient cells. These results indicate that hSMG-1 plays important roles in the maintenance of both genome and transcriptome integrity in human cells.
PMID:15175154 | DOI:10.1016/j.molcel.2004.05.005
View details for PubMedID 15175154
-
More
-
Direct regulation of CREB transcriptional activity by ATM in response to genotoxic stress Proceedings of the National Academy of Sciences of the United States of America
Shi Y, Venkataraman SL, Dodson GE, Mabb AM, LeBlanc S, Tibbetts RS
2004 Apr 20;101(16):5898-903. doi: 10.1073/pnas.0307718101. Epub 2004 Apr 8.
-
More
Ataxia-telangiectasia (A-T) is a syndrome of cancer susceptibility, immune dysfunction, and neurodegeneration that is caused by mutations in the A-T-mutated (ATM) gene. ATM has been implicated as a critical regulator of cellular responses to DNA damage, including the activation of cell cycle checkpoints and induction of apoptosis. Although defective cell cycle-checkpoint regulation and associated genomic instability presumably contribute to cancer susceptibility in A-T, the mechanism of neurodegeneration in A-T is not well understood. In addition, although ATM is required for the induction of the p53 transcriptional program in response to DNA damage, the identities of the relevant transcription factors that mediate ATM-dependent changes in gene expression remain largely undetermined. In this article, we describe a signal transduction pathway linking ATM directly to the Ca(2+)/cAMP response element-binding protein, CREB, a transcription factor that regulates cell growth, homeostasis, and survival. ATM phosphorylated CREB in vitro and in vivo in response to ionizing radiation (IR) and H(2)O(2) on a stress-inducible domain. IR-induced phosphorylation of CREB correlated with a decrease in CREB transactivation potential and reduced interaction between CREB and its transcriptional coactivator, CREB-binding protein (CBP). A CREB mutant containing Ala substitutions at ATM phosphorylation sites displayed enhanced transactivation potential, resistance to inhibition by IR, and increased binding to CBP. We propose that ATM-mediated phosphorylation of CREB in response to DNA damage modulates CREB-dependent gene expression and that dysregulation of the ATM-CREB pathway may contribute to neurodegeneration in A-T.
PMID:15073328 | PMC:PMC395895 | DOI:10.1073/pnas.0307718101
View details for PubMedID 15073328
-
More
-
ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses Nature
Bao S, Tibbetts RS, Brumbaugh KM, Fang Y, Richardson DA, Ali A, Chen SM, Abraham RT, Wang XF
2001 Jun 21;411(6840):969-74. doi: 10.1038/35082110.
-
More
Genotoxic stress triggers the activation of checkpoints that delay cell-cycle progression to allow for DNA repair. Studies in fission yeast implicate members of the Rad family of checkpoint proteins, which includes Rad17, Rad1, Rad9 and Hus1, as key early-response elements during the activation of both the DNA damage and replication checkpoints. Here we demonstrate a direct regulatory linkage between the human Rad17 homologue (hRad17) and the checkpoint kinases, ATM and ATR. Treatment of human cells with genotoxic agents induced ATM/ATR-dependent phosphorylation of hRad17 at Ser 635 and Ser 645. Overexpression of a hRad17 mutant (hRad17AA) bearing Ala substitutions at both phosphorylation sites abrogated the DNA-damage-induced G2 checkpoint, and sensitized human fibroblasts to genotoxic stress. In contrast to wild-type hRad17, the hRad17AA mutant showed no ionizing-radiation-inducible association with hRad1, a component of the hRad1-hRad9-hHus1 checkpoint complex. These findings demonstrate that ATR/ATM-dependent phosphorylation of hRad17 is a critical early event during checkpoint signalling in DNA-damaged cells.
PMID:11418864 | DOI:10.1038/35082110
View details for PubMedID 11418864
-
More
-
Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress Genes & development
Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ, Abraham RT
2000 Dec 1;14(23):2989-3002. doi: 10.1101/gad.851000.
-
More
The BRCA1 gene encodes a tumor suppressor that is mutated in 50% of familial breast cancers. The BRCA1 protein has been implicated in the DNA damage response, as DNA damage induces the phosphorylation of BRCA1 and causes its recruitment into nuclear foci that contain DNA repair proteins. The ataxia-telangiectasia-mutated (ATM) gene product controls overall BRCA1 phosphorylation in response to gamma-irradiation (IR). In this study, we show that BRCA1 phosphorylation is only partially ATM dependent in response to IR and ATM independent in response to treatment with UV light, or the DNA replication inhibitors hydroxyurea (HU) and aphidicolin (APH). We provide evidence that the kinase responsible for this phosphorylation is the ATM-related kinase, ATR. ATR phosphorylates BRCA1 on six Ser/Thr residues, including Ser 1423, in vitro. Increased expression of ATR enhanced the phosphorylation of BRCA1 on Ser 1423 following cellular exposure to HU or UV light, whereas doxycycline-induced expression of a kinase-inactive ATR mutant protein inhibited HU- or UV light-induced Ser 1423 phosphorylation in GM847 fibroblasts, and partially suppressed the phosphorylation of this site in response to IR. Thus, ATR, like ATM, controls BRCA1 phosphorylation in vivo. Although ATR isolated from DNA-damaged cells does not show enhanced kinase activity in vitro, we found that ATR responds to DNA damage and replication blocks by forming distinct nuclear foci at the sites of stalled replication forks. Furthermore, ATR nuclear foci overlap with the nuclear foci formed by BRCA1. The dramatic relocalization of ATR in response to DNA damage points to a possible mechanism for its ability to enhance the phosphorylation of substrates in response to DNA damage. Together, these results demonstrate that ATR and BRCA1 are components of the same genotoxic stress-responsive pathway, and that ATR directly phosphorylates BRCA1 in response to damaged DNA or stalled DNA replication.
PMID:11114888 | PMC:PMC317107 | DOI:10.1101/gad.851000
View details for PubMedID 11114888
-
More
-
Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine Cancer research
Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT
1999 Sep 1;59(17):4375-82.
-
More
Caffeine exposure sensitizes tumor cells to ionizing radiation and other genotoxic agents. The radiosensitizing effects of caffeine are associated with the disruption of multiple DNA damage-responsive cell cycle checkpoints. The similarity of these checkpoint defects to those seen in ataxia-telangiectasia (A-T) suggested that caffeine might inhibit one or more components in an A-T mutated (ATM)-dependent checkpoint pathway in DNA-damaged cells. We now show that caffeine inhibits the catalytic activity of both ATM and the related kinase, ATM and Rad3-related (ATR), at drug concentrations similar to those that induce radiosensitization. Moreover, like ATM-deficient cells, caffeine-treated A549 lung carcinoma cells irradiated in G2 fail to arrest progression into mitosis, and S-phase-irradiated cells exhibit radioresistant DNA synthesis. Similar concentrations of caffeine also inhibit gamma- and UV radiation-induced phosphorylation of p53 on Ser15, a modification that may be directly mediated by the ATM and ATR kinases. DNA-dependent protein kinase, another ATM-related protein involved in DNA damage repair, was resistant to the inhibitory effects of caffeine. Likewise, the catalytic activity of the G2 checkpoint kinase, hChk1, was only marginally suppressed by caffeine but was inhibited potently by the structurally distinct radiosensitizer, UCN-01. These data suggest that the radiosensitizing effects of caffeine are related to inhibition of the protein kinase activities of ATM and ATR and that both proteins are relevant targets for the development of novel anticancer agents.
PMID:10485486
View details for PubMedID 10485486
-
More
-
A role for ATR in the DNA damage-induced phosphorylation of p53 Genes & development
Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT
1999 Jan 15;13(2):152-7. doi: 10.1101/gad.13.2.152.
-
More
Phosphorylation at Ser-15 may be a critical event in the up-regulation and functional activation of p53 during cellular stress. In this report we provide evidence that the ATM-Rad3-related protein ATR regulates phosphorylation of Ser-15 in DNA-damaged cells. Overexpression of catalytically inactive ATR (ATRki) in human fibroblasts inhibited Ser-15 phosphorylation in response to gamma-irradiation and UV light. In gamma-irradiated cells, ATRki expression selectively interfered with late-phase Ser-15 phosphorylation, whereas ATRki blocked UV-induced Ser-15 phosphorylation in a time-independent manner. ATR phosphorylated p53 at Ser-15 and Ser-37 in vitro, suggesting that p53 is a target for phosphorylation by ATR in DNA-damaged cells.
PMID:9925639 | PMC:PMC316393 | DOI:10.1101/gad.13.2.152
View details for PubMedID 9925639
-
More
-
Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin Cancer research
Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT
1998 Oct 1;58(19):4375-82.
-
More
Members of the phosphatidylinositol-3 kinase related kinase (PIKK) family function in both cell cycle progression and DNA damage-induced cell cycle checkpoints. The fungal metabolite, wortmannin, is an effective radiosensitizer that irreversibly inhibits certain members of the PIKK family. Based on their roles in DNA damage responses, several PIKKs, DNA-dependent protein kinase (DNA-PK), ataxia telangiectasia mutated (ATM) and the ataxia- and Rad3-related protein (ATR), are potential targets for the radiosensitizing effect of wortmannin. In this report, we demonstrate that wortmannin is a relatively potent inhibitor of DNA-PK (IC50, 16 nM) and ATM (IC50, 150 nM) activities, whereas ATR activity is significantly less sensitive to this drug (IC50, 1.8 microM). In intact A549 lung adenocarcinoma cells, wortmannin inhibited both DNA-PK and ATM at concentrations that correlated closely with those required for radiosensitization. Furthermore, pretreatment of A549 cells with wortmannin resulted in radioresistant DNA synthesis, a characteristic abnormality of ATM-deficient cells. These results identify wortmannin as an inhibitor of ATM activity and suggest that ATM and DNA-PK are relevant targets for the radiosensitizing effect of this drug in cancer cells.
PMID:9766667
View details for PubMedID 9766667
-
More
-
TcDJ1, a putative mitochondrial DnaJ protein in Trypanosoma cruzi FEMS microbiology letters
Carreira MA, Tibbetts RS, Olson CL, Schuster C, Renz M, Engman DM, Goldenberg S
1998 Sep 1;166(1):141-6. doi: 10.1111/j.1574-6968.1998.tb13195.x.
-
More
A full length cDNA encoding a novel Trypanosoma cruzi DnaJ protein was cloned and characterized. The 324 amino acid protein encoded by the cDNA (TcDJ1) displays a characteristics J-domain, but lacks the Gly-Phe and zinc finger regions present in some other DnaJ proteins. Relative to four other T. cruzi DnaJ proteins, TcDJ1 has an amino terminal extension containing basic and hydroxylated resides characteristic of mitochondrial import peptides. A T. cruzi transfectant expressing epitope-tagged TcDJ1 was generated and subcellular fractions were produced. Western blot analysis revealed that the protein has a molecular mass of 29 kDa and is found in the mitochondrial fraction. The expression of TcDJ1 is developmentally regulated since the levels of both mRNA and protein are much higher in epimastigotes (replicative form) than in metacyclic trypomastigotes (infective form). Thus it may participate in mitochondrial biosynthetic processes in this organism.
PMID:9741092 | DOI:10.1111/j.1574-6968.1998.tb13195.x
View details for PubMedID 9741092
-
More
-
The DnaJ family of protein chaperones in Trypanosoma cruzi Molecular and biochemical parasitology
Tibbetts RS, Jensen JL, Olson CL, Wang FD, Engman DM
1998 Mar 15;91(2):319-26. doi: 10.1016/s0166-6851(97)00214-4.
-
More
We have molecularly cloned four members of the DnaJ (heat shock protein 40) family of protein chaperones of the protozoan parasite Trypanosoma cruzi--tcj1, tcj2, tcj3 and tcj4. While all the proteins contain defining J domains at their N-termini, only tcj2, tcj3 and tcj4 contain glycine/phenylalanine-rich and zinc finger domains common to many other DnaJ homologues. Furthermore, tcj2 and tcj4 contain C-terminal CaaX motifs, substrates for prenyl modifications, suggesting that they are associated with cellular membranes. tcj1 is a divergent member of the family, containing neither glycine/phenylalanine-rich nor zinc finger domains. All the T. cruzi DnaJ genes are single copy, in contrast to other T. cruzi heat shock genes, which are arranged in multicopy direct tandem arrays. Among the tcj mRNAs, only tcj2 is heat inducible, which may result from posttranscriptional regulation involving a sequence found in the 3' untranslated regions of all heat-inducible T. cruzi mRNAs described to date. Further study of this important family of protein chaperones will aid our understanding of the protein folding and assembly processes in protozoans.
PMID:9566524 | DOI:10.1016/s0166-6851(97)00214-4
View details for PubMedID 9566524
-
More
-
Cardiac antigen-specific autoantibody production is associated with cardiomyopathy in Trypanosoma cruzi-infected mice Journal of immunology (Baltimore, Md. : 1950)
Tibbetts RS, McCormick TS, Rowland EC, Miller SD, Engman DM
1994 Feb 1;152(3):1493-9.
-
More
An inflammatory cardiomyopathy may develop in humans and experimental animals with chronic Trypanosoma cruzi infection (Chagas' disease). Among the possible mechanisms involved in the pathogenesis of Chagas' cardiomyopathy, induction of heart-specific autoimmune responses has recently received substantial experimental support. The goal of the current study was to determine whether cardiac Ag-specific antibodies are produced in T. cruzi-infected mice with heart disease and, if so, to determine their Ag specificities. Upon infection with the Brazil strain of T. cruzi, C57BL/6 mice develop a cardiomyopathy that is histologically similar to that observed in chronically infected humans. Antisera from these mice were found to react with three cardiac Ag, having relative molecular masses of 200, 150, and 53 kDa. p200 and p150 are specifically found in heart muscle, although p53 is found in skeletal muscle as well. C57BL/6 mice infected with the Guayas strain of T. cruzi, which do not develop cardiomyopathy, did not produce antibodies to p200, p150, or p53, indicating that these antibodies may be specific markers of cardiomyopathy. Finally, p200 and p53 were identified as the contractile protein myosin and the intermediate filament protein desmin, respectively. This last finding is of special interest, because antibodies specific for myosin or desmin have been detected in humans and experimental animals with other natural and experimental cardiomyopathies. This suggests that infection with particular strains of T. cruzi may lead to the development of a cardiac Ag-specific autoimmune disease, possibly involving one or more of the Ag identified in this study.
PMID:8301148
View details for PubMedID 8301148
-
More
-
Molecular cloning and characterization of the 78-kilodalton glucose-regulated protein of Trypanosoma cruzi Infection and immunity
Tibbetts RS, Kim IY, Olson CL, Barthel LM, Sullivan MA, Winquist AG, Miller SD, Engman DM
1994 Jun;62(6):2499-507. doi: 10.1128/iai.62.6.2499-2507.1994.
-
More
The protozoan Trypanosoma cruzi is the etiologic agent of Chagas' disease, an illness responsible for morbidity and death among millions of Latin Americans. Mice also develop this disease when infected with T. cruzi and are a useful model organism for the study of parasite-specific immune responses. To identify immunogenic T. cruzi antigens, serum from an infected mouse was used to isolate clones from a T. cruzi epimastigote cDNA expression library. One of these clones was found to encode the 78-kDa glucose-regulated protein (grp78), the endoplasmic reticular member of the 70-kDa heat shock protein (hsp70) family. Like the mammalian and yeast grp78s, the T. cruzi protein contains an endoplasmic reticular leader peptide and a carboxyl-terminal endoplasmic reticular retention sequence. T. cruzi grp78 is encoded by a tandemly arranged family of three genes located on a chromosome of 1.6 Mb. The effects on grp78 expression of heat shock and tunicamycin treatment, the latter of which specifically stimulates mammalian grp78, were investigated. While the level of the grp78 protein remained constant under all circumstances, grp78 mRNA was unaffected by heat shock but induced fivefold by tunicamycin. Finally, we found that grp78 is the most immunogenic of the T. cruzi heat shock proteins we have characterized, reacting strongly in immunoblots with sera from infected mice.
PMID:8188375 | PMC:PMC186537 | DOI:10.1128/iai.62.6.2499-2507.1994
View details for PubMedID 8188375
-
More
-
Trypanosoma cruzi heat-shock protein 90 can functionally complement yeast Molecular and biochemical parasitology
Palmer G, Louvion JF, Tibbetts RS, Engman DM, Picard D
1995 Mar;70(1-2):199-202. doi: 10.1016/0166-6851(95)00007-n.
-
Utility of recombinant flagellar calcium-binding protein for serodiagnosis of Trypanosoma cruzi infection Journal of clinical microbiology
Godsel LM, Tibbetts RS, Olson CL, Chaudoir BM, Engman DM
1995 Aug;33(8):2082-5. doi: 10.1128/jcm.33.8.2082-2085.1995.
-
More
The protozoan Trypanosoma cruzi is the causative agent of Chagas' disease, a major public health problem in Latin America and of growing concern in the United States as the number of infected immigrants increases. There is currently no testing of U.S. blood products for T. cruzi infection, and the best tests available, although highly sensitive, are not of high enough specificity to be useful for widespread screening of the blood supply in this country. Among the parasite antigens detected by sera of infected humans and mice, those in the range of 24 to 26 kDa are particularly reactive. With an aim of developing a sensitive, specific, recombinant antigen-based serologic test for T. cruzi infection, we used two antibody reagents specific for these 24- to 26-kDa antigens to isolate cDNA clones from a T. cruzi expression library. One clone was found to encode a previously characterized T. cruzi antigen, a 24-kDa flagellar calcium-binding protein (FCaBP). Recombinant FCaBP was found to be a sensitive, specific reagent for distinguishing T. cruzi-infected individuals from uninfected persons, and it therefore could potentially be used for screening purposes, especially if combined with other recombinant T. cruzi antigens that have similarly high degrees of diagnostic sensitivity and specificity.
PMID:7559952 | PMC:PMC228339 | DOI:10.1128/jcm.33.8.2082-2085.1995
View details for PubMedID 7559952
-
More
-
A rapid method for protein localization in trypanosomes Experimental parasitology
Tibbetts RS, Klein KG, Engman DM
1995 May;80(3):572-4. doi: 10.1006/expr.1995.1071.
Contact Information
Randal Tibbetts, PhD
3059 WIMR,1111 Highland Avenue
Madison, WI 53705